ALUMNOS CON DISCAPACIDAD VISUAL.  NECESIDADES Y RESPUESTA EDUCATIVA http://educacion.once.es/appdocumentos/educa/prod/Necesidades%20y%20respuesta%20educativa.pdf
NECESIDADES Y RESPUESTA EDUCATIVA http://educacion.once.es/appdocumentos/educa/prod/Necesidades%20y%20respuesta%20educativa.pdf
1. ¿QUÉ ES UNA DEFICIENCIA VISUAL?
Partiremos de la definición dada por la Organización Mundial de la salud (O.M.S.) según la cual una deficiencia es toda pérdida o anormalidad en una estructura a nivel fisiológico, anatómico o psicológico.
Por tanto, una deficiencia visual o ceguera se define funcionalmente, como la pérdida total o parcial del sentido de la vista. Hay que poner un límite a partir del cual se pueda considerar a una persona ciega o no, cuando se deja de ser considerada como vidente. Hay que añadir una dificultad: que estas personas en su mayoría van a tener un resto de visión, aunque éste sólo les permita ver unos determinados grados de luz, movimientos y sombras de objetos.
La O.M.S. va a incluir en este grupo a aquellas personas que tengan deficiencia del órgano de la visión.
Para determinar ese grado de ceguera actualmente se están utilizando dos parámetros, que son:
1. La agudeza visual: que es la capacidad para discriminar claramente los detalles finos en objetos que están situados a una distancia determinada. La agudeza visual va a ser el resultado de dividir la distancia a la que una persona ve un objeto por la distancia a la que tendría que ver si su visión fuese la adecuada. Este parámetro sólo informa de la visión foveal, que es la zona de la retina que nos informa de la mayor capacidad de agudeza visual, con lo cual este parámetro sólo va a determinar un tipo de deficiencia.a Escala de Wecker es la que utiliza la O.N.C.E.1y la O.M.S. para determinar la distancia del objeto.
2. Campo de visión: hace referencia a los límites de captación luminosa que tienen ambos ojos. Es decir, qué capacidad tiene el ojo para recibir la información luminosa que le llega desde distintos ángulos.
Los ángulos normales de visión son:
- por la parte externa o temporal la luz nos tiene que entrar en un ángulo de 90º.
- por la parte interna o nasal un ángulo de 60º.
- en la parte superior, el ángulo es de 50º
- en la parte inferior, el ángulo es de 70º
Organización Nacional de Ciegos Españoles.
Para la O.M.S. se considera ciego a aquella persona que no consigue tener en sus dos ojos, ni siquiera con corrección de cristales, una agudeza visual de 1/10 en la escala Wecker o una reducción en su campo de visión total del 35%.Para la O.N.C.E. se considera ciega a aquella persona que no conserva en ninguno de
sus dos ojos una agudeza visual menor al 1/10, o, no consigue contar los dedos de la mano a una distancia de tres metros. También a aquella persona que tiene una reducción en su campo visual igual o inferior al 10%.A nivel educativo conviene que los profesionales diferencien entre niños con deficiencia visual, niños con ceguera y niños con restos de visión. Aunque para el M.E.C.D. el tratamiento legal sea el mismo desdeel punto de vista funcional van a tener que trabajar de forma distinta.
2. CLASIFICACIÓN
La clasificación más utilizada en el campo educativo es la que realiza Barraga2, la cual divide a las personas con deficiencia visual en cuatro dimensiones:
Ciegos: aquellas personas que tienen visión nula o que únicamente puede percibir algunas gradaciones de luz. Desde el punto de vista educativo no van a poder utilizar la visión para adquirir ningún conocimiento, por locual todos esos conocimientos se van a basar en la utilización del braille.
Ciegos parciales: aquellas persona que tienen un resto visual que les permite percibir la luz, algunas gradaciones de color, pueden distinguir bultos y contornos. Pero la visión funcional que tiene va a ser muy reducida ( igual o menor 1/10). Barraga los diferencia porque a pesar de que la mayoría de los conocimientos los adquieren a través del Braille, va a ser fundamental que se intente potenciar el resto de visión.
Baja visión: son personas con un resto visual que les permite ver objetos a pocos centímetros. Barraga recomienda que a estas personas se les enseñe a desenvolverse cuanto antes en el sistema braille. Dice también que los centros educativos deben de compromenterse a dotarles de los recursos necesarios para que la mayoría de los conocimientos sean aprendidos a través de su resto visual ( suelen ser alumnos de
integración).
Personas con limitación visual: son personas con un mayor resto visual pero que, debido a su deficiencia, necesitan constantemente una iluminación adecuada, utilización de herramientas que les permitan acceder a los textos como lupas, Barraga, N. (1985) disminuidos visuales y aprendizaje. Enfoque evolutivo.
Barraga recomienda que, para llevar a cabo el tratamiento educativo, los profesionales han de conocer:
- el tipo de trastorno
- la etiología
- la gravedad del trastorno
- la evolución
- el pronóstico
- el origen
- el momento de aparición.
3. ORIGEN Y CARACTERÍSTICAS DE LA DEFICIENCIA VISUAL
Se ha comprobado que el origen puede ser múltiple: hereditario, congénito, vírico, traumático, recurrente a otra enfermedad o consecuencia de un proceso degenerativo por la edad.Como consecuencia de esto van a ser múltiples los factores que van a incidir en la capacidad de visión de esas personas. Eso implica, que dos personas con la misma alteración y con el mismo grado de deficiencia visual, puede presentar una evolución visual completamente distinta. Por lo que es difícil hablar de características generales de esta población.
EL SISTEMA VISUAL
El sistema visual está formado por el ojo, las vías nerviosas y las estructuras que están implicadas dentro del sistema nervioso central que se engloban en el cortex visual.6La información luminosa que entra del exterior para llegar a formar una imagen visual debe llegar al cerebro y el ojo; va a ser el encargado de recibir delexterior esa información luminosa y de transmitirla. En ella va estar implicado:
Cornea: que es la parte exterior del ojo, es casi circular y transparente. Su principal función es proteger todo el contenido interior del ojo y permite a su vez el paso de la luz a través de ella. La cornea junto con el cristalino van a enfocar esa información luminosa que llega del exterior en la retina. A su vez la cornea permite el 65% de refracción del ojo, es decir, cambiar el sentido de la dirección de la información que
entra del exterior.
Humor acuoso: éste se sitúa justo detrás de la cornea. Se caracteriza por se líquido y está drenando continuamente al ojo. Su función principal es la de eliminar las sustancias de desecho que se pueden acumular entre la cornea y el cristalino y otra es mantener la estructuras del ojo.
Iris: es la parte coloreada del ojo y permite que entre una mayor o menor cantidad de luz y eso lo va a hacer a través de la pupila. Por lo tanto, su función principal es regular el tamaño de la pupila.
Cristalino: éste es realmente la lente del ojo y se caracteriza por ser avascular, viconvexa, transparente, tiene un 65% de agua y un 35% de proteínas. Se mantiene suspenso justo detrás del iris gracias al cuerpo ciliar. Junto con la cornea va a ser la lente que enfoque la información luminosa en la retina.Con el paso de los años, ese cristalino va a ir aumentando de tamaño y perdiendo elasticidad ( capacidad de
acomodación). Esa es la principal causa de la presbicia o vista cansada.
Humor vítreo: se caracteriza por ser claro, gelatinoso y comprende 2/3 del volumen del ojo. Su función principal es la de mantener la transparencia y la estructura del globo ocular. Si este humor vítreo se infecta se va a volver nebuloso con lo cual se va a ver reducida significativamente la agudeza visual; si por el contrario sufrimos un traumatismo el ojo se hunde.
Esclerótica: que es la capa externa del ojo y recubre todo el globo ocular a excepción
de la cornea. La función principal de esta capa es la de proteger el contenido interno
del ojo de posibles infecciones.
Coroides: es la capa que se sitúa entre la esclerótica y la retina. Está compuesta de vasos sanguíneos con lo cual su principal función es aportar sangre al ojo y agua.
Retina: es la capa más interna y se la conoce como la zona sensible a la información luminosa. Es decir, que la retina va a recibir la información luminosa del exterior y gracias a su tejido neuronal va a transmitir esa información nerviosa que llega del cortex visual a través del nervio óptico.
Dentro de la retina tenemos que destacar cuatro componentes:
1-Conos y bastones: se denominan receptores de la información luminosa. Los conos van a intervenir en la visión del ojo y en la percepción de los colores. Se sitúan en las zonas centrales de la retina y en la fóvea. Son muy escasos.
2- Los bastones: se sitúan por toda la retina, excepto en la fóvea y en el punto ciego (que es el punto donde se une la retina con el nervio óptico); se caracterizan por ser muy sensibles a la luz (en situaciones de poco luz lo que nos permite ver son los bastones). También se encargan de que podamos distinguir los distintos grados de gris.
3- La fóvea y la mácula: son dos puntos que se sitúan en la zona central de la retina. La fóvea se la conoce por ser la zona de mayor sensibilidad visual, es decir, la zona donde se va a determinar la agudeza visual máxima de la persona. La mácula permite que la visión global sea la más óptima posible.
Nervio óptico: tiene como función transmitir los impulsos nerviosos directamente del cortex visual. Aquí se empieza a formar la imagen que percibe la persona, pero el que
realmente ve es el cerebro.
Conjuntiva: es una capa protectora del ojo.
Cuerpo ciliar: es el cuerpo que sostiene al cristalino.
EL MECANISMO DE LA VISIÓN: ¿CÓMO SE VE?
La visión va a depender de la información luminosa que atraviesen la cornea, humor acuoso, iris, cristalino, humor vitreo y la retina. A partir de hay, los impulsos nervioso se van a transmitir los impulsos hasta el cortex visual.Los impulsos nerviosos cuando pasan el nervio óptico se dirigen al quiasma a través de dos fibras: una interna y otra externa. Por lo tanto es en el quiasma donde se va a juntar la información proveniente de ambos ojos (el derecho y el izquierdo); de aquí se dirigen a las cintillas ópticas y pasando después a los cuerpos geniculados laterales. De estos cuerpos surgen una serie de ramificaciones nerviosas que se van a dirigir a los centros optomotores y centros pretectales.
Estos centros optomotores y pretectales cumplen tres funciones:
1-Permiten la acomodación del cristalino
2- Los movimientos oculares del ojo: arriba - abajo y derecha - izquierda.
3-Reflejo pupilar.
De aquí pasan a través de los tractos geniculocalcarinos y a la corteza cerebral ( zona occipital de la corteza cerebral o visual). Una vez que la información llega a la corteza para que se produzca una correcta visión, ésta tiene que pasar por tres áreas.
Éstas áreas son:
1- Área de proyección visual (área primaria): si se daña esta zona sólo percibirá luz.
2- Áreas secundarias
3- Áreas terciarias
Las dos últimas áreas se denominan asociativas y van a permitir identificar y discriminar formas, contornos y reconocer cambios en la información luminosa. Si se lesionan estas dos áreas las personas van a ser capaces de percibir luz pero les queda afectada la percepción del espacio, es decir, la agudeza visual.Actualmente para detectar las posibles lesiones que se pueden producir desde el nervio óptico hasta la corteza se utilizan los potenciales evocados.
TRASTORNOS DE LA VISIÓN
Los trastornos de la visión los vamos a encontrar en torno a tres grupos:
1- Alteraciones funcionales.9
2- Alteraciones estructurales.
3- Alteraciones de las vías nerviosas.
A) LAS ALTERACIONES FUNCIONALES MÁS GENERALES SON:
1- Disminución de la agudeza central o visión fluctuante. Se caracteriza por visión borrosa, dificultades para percibir la luz ( sensaciones de oscuridad constante) y la visión nebulosa.
2- La metamorfosia, presenta una dificultad para diferenciar contornos y visión nebulosa constante. Se va a caracterizar por ver los objetos convados y dificultad para ver el límite de esos objetos.
3- La Fotofobia es la incapacidad del ojo para adaptarse a las distintas gradaciones de luz y a los cambios de intensidad.
4- Discriminación de los colores: el sujeto puede tener problemas para identificar algunas o todas las gamas y para distinguir las tonalidades.
5- Deficiencias de campo: son ausencias de visión en determinadas partes del campo visual.
6- La ceguera nocturna es la incapacidad de distinguir objetos en situaciones de oscuridad.
7- Las imágenes enteptópicas se caracterizan por la aparición de manchas o elementos flotantes que entran en el campo visual y que pueden llegar a fijarse en la visión.
B) ALTERACIONES ESTRUCTURALES
1- LA CÓRNEA SE PUEDE LESIONAR DE DOS FORMAS:
Queratitis: se caracteriza por una inflamación de la cornea que se debe a una infección. Los síntomas son: fotofovea y dolor intenso. Pudiendo ser necesario un trasplante de cornea.
Queratocono: alteraciones en la estructura cornea que producen un estiramiento y
adquiere la forma de icono.Se reduce el campo visual y progresivamente se va perdiendo la agudeza visual. Las manifestaciones hereditarias suelen aparecer a partir de los 20 años.
2- ENFERMEDADES DEL HUMOR ACUOSO: 10
El humor acuoso aporta sustancias nutritivas, elimina desechos y contribuye a mantener la forma del ojo.
Glaucoma: producido por una alta presión intraocular que daña la retina y las fibras del nervio óptico, lo que conlleva cierta pérdida de visión y dureza del globo ocular. La pupila se torna verdosa.
El glaucoma puede conllevar pérdida de la agudeza visual, del campo visual, o ambas cosas, degenerando en ceguera total. Puede ser congénito ( adquirido antes del nacimiento) o aparecer en la edad adulta ( hereditario o consecuencia de un intervención quirúrgica). Puede ser tratada quirúrjicamente o con fármacos.
3- AFECCIONES DEL IRIS:
Aniridia: incapacidad del iris ( que controla la entrada de la luz) para desarrollarse plenamente, de manera que está ausente parcial o totalmente. Es un defecto hereditario y congénito. La apariencia es como si la pupila fuera sumamente grande. Suele conllevar fotofobia, disminución de la agudeza visual, cataratas, retina
infradesarrollada y nistagmo ( movimientos rápidos, involuntarios y repetidos de los ojos) que conllevan una visión imperfecta, disminución de la agudeza visual, movimientos ilusorios de objetos y vértigo.
4- ALTERACIONES DEL CRISTALINO:
Cataratas: consisten en una pérdida de su transparencia, se opacifica. No conlleva dolor, ya que carece de fibras dolorosas, vasos sanguíneos y nervios. Gran variedad en cuanto al grado de densidad y al origen aunque suele estar relacionado con el envejecimiento. La disminución de la agudeza visual suele ser directamente proporcional a la densidad de la catarata, pudiendo degenerar en ceguera. Las más
comunes son:
- Cataratas congénitas: presentes en el nacimiento o desarrolladas poco después del mismo. El 25% de las cosas suele ser hereditario. Si la deficiencia visual es grave, será necesaria una intervención quirúrgica. Puede ser consecuencia de: Síndrome Down, medicamentos en la gestación, desnutrición 11grave en el embarazo,Síndrome marfán (anomalía congénita hereditaria de los tejidos conjuntivos del cuerpo).
- Cataratas subcapsulares posteriores: la parte posterior del cristalino se pone nebulosa. El efecto sobre la visión puede ser negativo. Ven mal a la luz del sol y tienen escasa visión de cerca aunque los campos visuales suelen ser normales; también pueden apreciar una cierta alteración del color y experimentar deslumbramientos ( especialmente durante la noche) debido a la dispersión de la luz por la opacidad. Pueden ser seniles ( por la edad) o traumáticos (lesión).
5- ALTERACIONES EN EL HUMOR VÍTREO:
Centelleo vítreo o aparición de luces intermitentes: la persona percibe una franja de luz localizada o que se mueve intermitentemente en el campo de visión durante una fracción de segundo. Su existencia no tiene una explicación clara; puede estar causada por una rotura de la retina, un desprendimiento o una hemorragia del vítreo.
Fibroplasia retrolental: se produce por la formación de tejido conjuntivo embrionario detrás del cristalino, apareciendo vasos sanguíneos y formaciones fibrosas en el vítreo. Con frecuencia se producen hemorragias, llegándose en algunos casos aldesprendimiento total o parcial de la retina. Está originada por la administración de niveles elevados de oxígeno a los bebés prematuros, aunque en ocasiones se ha encontrado también en bebés de gestación normal. Afecta a la agudeza visual, miopía grave, cicatrices y desprendimiento de retina con pérdida de campo de visión y posible ceguera.
6- ALTERACIONES DE LA RETINA:
El tejido retiniano es tejido conjuntivo por lo tanto es muy sensible a las alteraciones del riego sanguíneo. Fla mayoría de ellas producen visión borrosa. Si queda afectada la zona macular la agudeza visual central será deficiente y habrá dificultades para leer y para distinguir objetos a distancia. Si queda afectada la porción periférica la visión lateral quedará alterada pero podrá leer.
Diabetes mellitus: es una de las principales causas de ceguera. Se debe a una afección causada por la carencia de insulina en sangre. Los niveles altos de azucar afectan a los ojos, los riñones, la piel y el sistema circulatorio. Uno de los síntomas más tempranos es la pérdida de la capacidad de acomodación y la alteración en la capacidad de refracción. Los vasos sanguíneos de la retina pueden producir hemorragias, y éstas pueden extenderse al vítreo. Puede conllevar un desprendimiento de retina. La deficiencia visual puede ser muy variada, llegando incluso a la ceguera total. La agudeza visual se verá afectada en función de las partes de la retina que queden dañadas. Este tipo de diabetes puede llegar a desarrollar un glaucoma.
Retinosis pegmentaria: es una degeneración de los receptores de la retina de origen genético. Los bastones se van destruyendo poco a poco y el resto de la retina se atrofia. El primer síntoma es la dificultad para ver de noche; posteriormente los campos visuales se van a ver reducidos hasta perderse la visión macular también suele aparecer la fotofobia. Para esta enfermedad no se conoce tratamiento médico.
Desprendimiento de retina: con frecuencia es consecuencia de otros trastornos. La retina se separa de la coroides debido a traumatismos o enfermedades oculares. Al no recibir nutrición alguna, la porción desprendida se atrofia y se desarrolla una zona ciega en el campo de visión.
La aparición de luces intermitentes acompañadas de dolor punzante son indicios significativos de que se ha porducido o se va a producir un desprendimiento. Si se afecta a la mácula, la agudeza visual puede disminuir, la retina se hinchará, se producirá micropsia ( los objetos apareces más pequeños cuando se miran con el ojo afectado) y se alterará la visión de los colores. El tratamiento consiste en una operación para intentar reconectar la retina, con ésta suele recuperarse la vista.
Toxoplasmosis: es una infección intraocular grave, producida por la exposición a un
pequeño organismo que se transmite a través del contacto con animales domésticos o mediante la ingestión de carne cruda que lo contenga.La toxoplasmosis congénita es mucho más grave ya que afecta al tejido nervioso y puede causar daños graves al celebro La afección ocular es mucho más común en los casos congénitos. Es una enfermedad que actualmente se controla a las mujeres embarazadas o que desean tener hijos, por el riesgo que conlleva al feto.Si afecta a la zona macular la agudeza visual es muy reducida, pudiendo desembocar en estrabismo, sobre todo en niños pequeños. Generalmente no es progresiva. El tratamiento consiste en ayudas ópticas
Degeneración macular senil: es una enfermedad común en las personas mayores de 65 años. Aunque también puede darse en personas jóvenes que la heredan como degeneración macular juvenil. Afecta fundamentalmente a los dos ojos, aunque en principio, es sólo uno el que presenta problemas importantes.
Esta enfermedad produce una ceguera en el centro del campo visual y la visión periférica permanece intacta; haciendo imposible la lectura. Entre el 5 - 20% de los casos crecen vasos sanguíneos nuevos debajo del área macular que gotean líquido dentro de la mácula, destruyendo los conos. La progresión es gradual pero el resultado final es un escotoma central denso. No desemboca en ceguera total, aún cuando la visión central pueda ser muy deficiente; pudiendo conllevar fotofobia y deslumbramiento. El tratamiento va a ser enseñar a los pacientes a fijar su mirada en torno a la manca ciega central, y la fotocoagulación con láser que es para las degeneraciones maculares graves, que detienen o reducen muchas de las fugas de los vasos sanguíneos anómalos.
Albinismo: es una afección congénita caracterizada por una tez clara, pelo rubio platino e iris y cejas de color claro; debido a la carencia de pigmento o a la incapacidad del cuerpo para producirlo.
Puede afectar a todas las estructuras pigmentarias o solamente una de ellas. Si afecta únicamente a los ojos se denomina albinismo ocular; es una alteración hereditaria ( rasgo recesivo ligado al sexo; los hombres la padecen y las mujeres la portan). Va acompañado de fotofobia grave; puede haber nistagmo debido a una mácula infradesarrollada. También suelen aparecer defectos de refacción y astigmatismo. Los campos visuales suelen ser normales o tienen una ligera reducción. El tratamiento puede conllevar la aplicación de lentillas para mejorar la agudeza visual; las lentillas de colores son a menudo eficaces para reducir el deslumbramiento, mejorar el defecto de refracción y reducir el nistagmo.
Acromatopsia o ceguera a los colores: es una alteración genética que afecta a la porción nerviosa del ojo encargada de discernirlos. La mayoría conservan una agudeza visual normal. Cuando la deficiencia en el sistema de conos es total va a conllevar una gran disminución de la agudeza visual, que no va a poder corregirse con lentes convencionales. La visión de cerca suele quedar menos afectada que la visión de lejos; también va a aparecer fotofobia y nistagmos que se ven reducidos notablemente cuando descienden los niveles de iluminación.
7- ALTERACIONES DE LAS VÍAS NERVIOSAS:
Son alteraciones que afectan a la transmisión del impulso nervioso hacia el cerebro.
Atrofia del nervio óptico: causada por multitud de enfermedades o por herencia. La cabeza del nervio óptico se vuelve pálida, produciendo pérdida de la agudeza visual y cambios en el campo visual pudiendo llegar a ceguera total. Casi nunca es posible su tratamiento a no ser que la causa sea detectada tempranamente y tratada con eficacia.Las lesiones que se producen antes del quiasma óptico suelen ser unilaterales afectando a un sólo ojo. Las posteriores dan lugar a afecciones de la misma parte del campo visual de ambos ojos. Las lesiones situadas en el quiasma generalmente causan alteraciones bitemporales. Cuanto más similares sean los defectos en los dos campos visuales, la lesión estará situada en una región más posterior. Por lo tanto es más probable que la mácula esté intacta y se conserve la agudeza visual.14
C) SÍNDROMES MÁS FRECUENTES ASOCIADOS A LA DEFICIENCIA VISUAL.
Rubeola congénita: el virus de la rubeola puede alterar la división y multiplicación de las células y causar cambiós cromosómicos responsables de determinadas deficiencias congénitas. El niño con rubeola suele tener defectos en los ojos, el oído y el corazón. El glaucoma congénito y las cataratas congénitas dan a los ojos una apariencia nebulosa, blanquecina, impidiendo examinar la parte posterior del ojo. Los ojos suelen ser muy pequeños (microftalmía). La opacidad de la córnea y las cataratas reducen la agudeza visual, y el glaucoma congénito provoca restricciones en el campo visual. Pueden presentarse nistagmo y estrabismo. La afección no es progresiva y por si misma no parece causar problemas de visión.El tratamiento no es muy eficaz. Aunque las cataratas y el glaucoma pueden tratarse por intervención quirúrgica, el pronóstico es negativo. En el cristalino va a seguir estando presente determinado virus, que puede producir una inflamación posterior, como consecuencia de la operación.
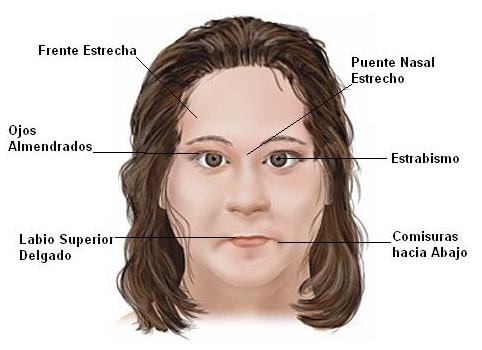
Síndrome de Down: es una afección congénita producida por una anomalía genética ( cromosoma 21 extra). Son generalmente personas con ojos estrechos y desviados, normalmente tienen retraso mental, con un cociente intelelectual entre 20 y 50. Suelen presentar problemas oculares, como nistagmo y miopía grave. El 50% presenta cataratas congénitas. El nivel de agudeza visual medible va a estar en función de su afección ocular y del nivel de inteligencia. Los campos visuales, la visión cromática y la visión de cerca suelen ser normales.
Síndrome de Marfan: es una anomalía congénita de los tejidos conjuntivos del cuerpo. Se cree que se transmite como rasgo dominante que afecta a ambos ojos por igual. Es hereditarias. Se caracterizan por tener dedos largos y finos en manos y pies, y alargamiento generalizando de las extremidades. Frecuentes problemas cardiovasculares y musculatura infradesarrollada. Conlleva muchas complicaciones oculares, siendo la más frecuente la luxación de cristalino (dislocación, visión borrosa generalizada). En ocasiones se puede dar la visión doble en uno o en ambos ojos. Conlleva una disminución de la agudeza visual.Pueden presentar también una pupila dislocada o múltiple. La miopía fuerte es común, desencovando en desprendimiento de retina. También es posible que se desarrollo el estrabismo, debido a la disminución de las funciones visuales. No se recomienda la intervención quirúrgica. Externamente puede aparecer heterocromía de iris (ojos de diferentes colores), esclerótica azulada y nistagmo.
3.1. DEFECTOS DE REFRACCIÓN
D)DEFECTOS DE REFRACCIÓN
HIPERMETROPÍA15
Es el estado de la refracción ocular en el que los rayos paralelos que inciden en el ojo se focalizan por detrás de la retina. Se puede clasificar la hipermetropía en tres tipos: latente, manifiesta y total.La agudeza visual puede variar según el grado de acomodación del paciente, pero si carece de acomodación la agudeza visual será mala de lejos y de cerca.Puede aparecer la astenopía acomodativa: la hipermetropía no corregida con cristales puede ser compensada con la acomodación, pero este esfuerzo conduce a la fatiga del músculo ciliar.
MIOPÍA
Es el estado de refracción ocular en el que los rayos que inciden en el ojo se enfocan por delante de la retina.
Hay dos tipos:
Miopía benigna o simple: El paciente tiene mala visión de lejos y el resto del examen ocular es normal.
Miopía maligna: Se inicia precozmente y aumenta a lo largo de toda la vida. En edades avanzadas el paciente puede llegar a la ceguera dado el carácter degenerativo de la enfermedad.
ASTIGMATISMO
Es un defecto de la refracción ocular en el que nunca se reúnen en un foco la totalidad de los rayos paralelos que atraviesan los medios dióptricos del ojo.