lunes, 27 de mayo de 2013
miércoles, 8 de mayo de 2013
MACROCEFALIA
Características
Es un defecto raro del desarrollo cerebral en el que el cerebro crece de forma excesiva durante los primeros meses de vida del bebé, y esto provoca un crecimiento anormalmente rápido del cráneo. La macrocefalia se define como el aumento del perímetro craneal en más de dos desviaciones estándar por encima de los rangos normales según las tablas de crecimiento craneal diferenciadas por sexo y edad, grupo étnico y edad. Es un término descriptivo más que de diagnóstico y es una característica de una variedad de trastornos, aunque si su hijo tiene sólo la cabeza grande, inclusive sobrepasando los límites normales pero su desarrollo en general es normal (puede controlar su cabeza, tiene buen tono muscular, tensión en los músculos que permite moverlos, mantener una posición, etc.); y si tiene predisposición familiar a tener cabeza grande, se trataría de una Macrocefalia familiar benigna.
Causas
El tamaño de la cabeza del bebé es una característica que debe ser cuidadosamente observada desde el nacimiento, y forma parte mediciones que debe realizar el pediatra. El tamaño se evalúa de acuerdo a tablas de crecimiento cefálico desde el nacimiento hasta los 18 años. El volumen de cada uno de los tres componentes del cráneo (cerebro, líquido cefalorraquídeo y sangre) determinan el tamaño de la cavidad craneal durante la infancia. El crecimiento exagerado de uno de estos tres componentes, se realiza a expensas de los otros dos para mantener constantes el volumen y la presión dentro del cráneo (intracraneal). Si la curva de crecimiento del perímetro cefálico tiende a salirse de los rangos normales, puede ser un síntoma de algún problema. Cuando el perímetro es menor se dice que hay Microcefalia y cuando es mayor, se dice que hay Macrocefalia. El crecimiento del cráneo está íntimamente relacionado con el crecimiento del cerebro y con la circulación del liquido cefalorraquídeo ; cualquier entidad que condicione un aumento del tamaño del cerebro o de la cantidad de liquido cefalorraquídeo, va a reflejarse en el tamaño del cráneo.
Diagnóstico
Perímetro craneal es importante porque ofrece información sobre el desarrollo del cerebro del bebé. En el recién nacido el perímetro cefálico puede estar disminuido por la presión producida al pasar por el canal del parto. Como sabemos, el cráneo de los bebés está formado por placas óseas móviles, no soldadas, que puedan montarse entre sí: las llamadas fontanelas. A veces es evidente que los huesos se han montado unos sobre otros, o puede haber alguna deformidad. La medida media del perímetro craneal al nacer es de 35 ó 36 centímetros y en los primeros años de vida experimenta un gran aumento debido al crecimiento del encéfalo y a la maduración del sistema nervioso central.
Para realizar el diagnóstico el pediatra medirá el perímetro craneal del bebé desde que nace hasta los tres años aproximadamente, para valorar si el crecimiento es normal o si ha aumentado significativamente. Sin embargo, en algunos casos una sola medición es suficiente para confirmar el incremento. El diagnóstico de la macrocefalia se hace al comprobar que la medida del perímetro craneal está por encima de la normalidad. (Es esencial medir el perímetro craneal de los padres y hermanos, para valorar los factores de la constitución familiar). El médico se basará en la historia clínica del bebé en la que anotará los antecedentes personales y familiares, los síntomas que presenta en ese momento, lo que observa al hacer el examen físico y los resultados de las pruebas de laboratorio y de imagen complementarias.
MICROCEFALIA
Es una afección en la cual la cabeza de una persona es considerablemente más pequeña de lo normal para su edad y sexo, con base en tablas de referencia. El tamaño de la cabeza se mide como la distancia alrededor de la parte superior de la cabeza.
Causas
La microcefalia se presenta con mayor frecuencia debido a que el cerebro no logra crecer a una tasa normal. El crecimiento del cráneo está determinado por el del cerebro, el cual tiene lugar en el útero y durante la lactancia.
Las enfermedades que afectan el crecimiento cerebral pueden ocasionar microcefalia. Esto incluye infecciones, trastornos genéticos y desnutrición grave.
Los trastornos genéticos que causan microcefalia abarcan:
- Síndrome de Cornelia de Lange
- Síndrome del maullido de gato
- Síndrome de Down
- Síndrome de Rubinstein-Taybi
- Síndrome de Seckel
- Síndrome Smith-Lemli-Opitz
- Trisomía 18
- Trisomía 21
Estas otras afecciones pueden indirectamente causar microcefalia:
- Fenilcetonuria (FC) materna no controlada
- Intoxicación con metilmercurio
- Rubéola congénita
- Toxoplasmosis congénita
- Citomegalovirus congénito (CMV)
- Uso de ciertos fármacos durante el embarazo, especialmente alcohol y fenitoína
CRETINISMO
El cretinismo
Es una enfermedad que aparece en las primeras etapas de la vida, y que se produce por un defecto en la fabricación de hormona tiroidea durante la vida fetal.
Esto sucede generalmente en casos de ausencia o malformación de la glándula tiroides (glándula situada en la parte anterior y superior de la tráquea, cuyas hormonas influyen en el metabolismo y en el crecimiento), o en ciertos trastornos congénitos (que suceden antes de nacer) del tiroides que se ve incapaz para fabricar la hormona tiroidea.
Esto sucede generalmente en casos de ausencia o malformación de la glándula tiroides (glándula situada en la parte anterior y superior de la tráquea, cuyas hormonas influyen en el metabolismo y en el crecimiento), o en ciertos trastornos congénitos (que suceden antes de nacer) del tiroides que se ve incapaz para fabricar la hormona tiroidea.
Excepcionalmente se observa en poblaciones donde existe una carencia grave de yodo en la dieta.
El hipotiroidismo juvenil es un trastorno similar al anterior que se desarrolla en niños que aparentemente eran con anterioridad normales. Generalmente se produce por una inflamación aguda del tiroides, aunque en algunos casos se encuentran también trastornos en el metabolismo del yodo (utilización del yodo para fabricar la tiroxina) en esta glándula.
En los niños cretinos, el tiroides puede estar ausente o encontrarse reducido a un tamaño testimonial, sin que se pueda encontrar tejido glandular capaz de funcionar y de segregar la hormona tiroidea, tras haber sido sustituido por un tejido fibroso e inflamatorio (especie de tejido cicatricial que no tiene actividad).
En algunas familias en las que existen trastornos congénitos de la síntesis o fabricación de la hormona tiroidea, se encuentran bocios grandes (aumento del tamaño de la glándula tiroides), y en los estudios al microscopio se observa que la arquitectura del tejido de la glándula, está completamente desestructurada.
Como consecuencia de todo esto, el desarrollo físico y mental del niño está retrasado, así como el crecimiento de los huesos, de los dientes y del cerebro en su conjunto.
Algunos casos son heredados y éstos, presentan distintos trastornos del metabolismo del yodo y de la fabricación de la hormona tiroidea, imprescindible para el normal funcionamiento del organismo.
Síntomas
En los niños con cretinismo es característica la presentación de una piel gruesa, seca, arrugada y de un color amarillento y fría.
El aumento del tamaño de la lengua que se sale de la boca es un rasgo muy característico de estos niños que presentan unos labios engrosados en una boca entreabierta con babeo permanente.
El aumento del tamaño de la lengua que se sale de la boca es un rasgo muy característico de estos niños que presentan unos labios engrosados en una boca entreabierta con babeo permanente.
Los rasgos de la cara son característicos, siendo ésta muy ancha, con una nariz corta y respingona hacia arriba a la vez que chata debajo de una frente arrugada.
La observación de las manos y los pies muestran unas extremidades hinchadas en forma de pala. Es muy característica también la presentación de una hernia en el ombligo y un vientre muy abultado.
Desde el punto de vista psicológico, estos niños se encuentran embotados y apáticos y, generalmente, tienen un mal apetito y un tránsito intestinal habitualmente estreñido. Como consecuencia de la falta de la hormona tiroidea, la frecuencia cardiaca es lenta y la temperatura corporal baja.
Los niños cretinos pueden presentar al nacer un tamaño y peso normal o incluso más alto de lo habitual, no obstante el problema metabólico derivado de la ausencia o deficiencia de hormona tiroidea va a condicionar un crecimiento lento para su edad, haciendo que de adultos tengan una talla baja o incluso enana.
Es bastante característico del cretinismo el retraso mental que puede ser variable desde un retraso discreto a una minusvalía intensa.
Cuando el hipotiroidismo se desarrolla en la infancia, no son cretinos de nacimiento, los niños pueden aparecer clínicamente normales pero refieren cansancio, dificultad para la concentración, etc.
Cuando el hipotiroidismo se desarrolla en la infancia, no son cretinos de nacimiento, los niños pueden aparecer clínicamente normales pero refieren cansancio, dificultad para la concentración, etc.
Diagnóstico
En el diagnóstico de esta enfermedad es muy importante la sospecha al explorar el niño cuando nace. Unos meses después del nacimiento es característico el aspecto del niño que tiene una conducta letárgica y rasgos físicos similares a los descritos en el apartado anterior.
Cuando se sospecha el diagnóstico, se puede confirmar mediante la realización de análisis en sangre en los que se puede determinar la hormona tiroidea, la TSH (hormona segregada en la hipófisis, glándula endocrina muy pequeña, situada en la parte anteroinferior del encéfalo, que estimula al tiroides, llamada hormona tirotropa) y la determinación de yodo en la sangre.
La realización a estos niños de una radiografía del esqueleto, pone de manifiesto los rasgos típicos del cretinismo.
En algunos casos pueden existir a pesar de todo dudas, estando indicado el tratamiento inmediato con tiroxina, que debe instaurarse lo antes posible para prevenir un daño cerebral irreversible. Cuando la respuesta es positiva, se continúa el tratamiento aunque sin renunciar a confirmar el diagnóstico de forma inequívoca.
La experiencia clínica de los médicos es muy importante para el diagnóstico y el manejo de esta enfermedad, que en algunos casos puede confundirse con el mongolismo, aunque estos pacientes tienen las pruebas de función del tiroides normales. Sin embargo, presentan un estudio de los cromosomas patológicos, lo que los diferencia nítidamente de los niños cretinos.
En el hipotiroidismo juvenil, donde los niños pueden tener un aspecto físico normal, y sobrevenir la enfermedad como consecuencia de una inflamación aguda del tiroides, es muy importante estudiar y realizar análisis de anticuerpos antitiroideos circulantes (prueba de laboratorio para detectar anticuerpos producidos por organismo contra su propio tiroides, causando su destrucción y provocando la denominada tiroiditis de Hashimoto).
PRONÓSTICO.
Respecto al pronóstico, el enanismo físico y mental una vez aparecido es irreversible. Sin embargo, si se descubre y se trata pronto es perfectamente evitable siendo el pronóstico del crecimiento y del desarrollo mental normal en la mayoría de los casos.
En el hipotiroidismo juvenil la recuperación debe ser completa con el tratamiento adecuado con hormona tiroidea.
En el hipotiroidismo juvenil la recuperación debe ser completa con el tratamiento adecuado con hormona tiroidea.
LA FENILCETONURIA
La fenilcetonuria
Es una rara afección en la cual un bebé nace sin la capacidad para descomponer apropiadamente un aminoácido llamado fenilalanina.
es una enfermedad hereditaria, lo cual significa que se transmite de padres a hijos. Ambos padres deben transmitir el gen defectuoso para que el bebé padezca la enfermedad, lo que se denomina un rasgo autosómico recesivo.
Los bebés con fenilcetonuria carecen de una enzima denominada fenilalanina hidroxilasa, necesaria para descomponer un aminoácido esencial, llamado fenilalanina, que se encuentra en alimentos que contienen proteína.
Sin la enzima, los niveles de fenilalanina y dos substancias estrechamente relacionadas se acumulan en el cuerpo. Estas sustancias son dañinas para el sistema nervioso central y ocasionan daño cerebral.
Síntomas
La fenilalanina juega un papel en la producción corporal de melanina, el pigmento responsable del color de la piel y del cabello. Por lo tanto, los niños con esta afección usualmente tienen un cutis, cabello y ojos más claros que sus hermanos o hermanas sin la enfermedad.
Otros síntomas pueden ser:
- Retraso de las habilidades mentales y sociales
- Tamaño de la cabeza considerablemente por debajo de lo normal
- Hiperactividad
- Movimientos espasmódicos de brazos y piernas
- Discapacidad intelectual
- Convulsiones
- Erupción cutánea
- Temblores
- Postura inusual de las manos
Si la afección se deja sin tratamiento o si no se evitan los alimentos que contienen fenilalanina, se puede detectar un olor "a ratón" o "a moho" en el aliento, la piel y la orina. Este olor inusual se debe a la acumulación de sustancias de fenilalanina en el cuerpo.
El síndrome X frágil
El síndrome X frágil
es la forma más común de retraso mental hereditario. La enfermedad es causada por un gen específico. Normalmente, el gen produce una proteína necesaria para el desarrollo cerebral. Pero la mutación hace que una persona produzca poco o nada de dicha proteína, lo que resulta en el síntoma de X frágil.
Las personas que tienen solamente un pequeño cambio en el gen no tiene síntomas de X frágil. Las personas con cambios mayores pueden tener síntomas severos. Los mismos pueden incluir:
- Problemas de inteligencia, que van desde las discapacidades en el aprendizaje hasta el retraso mental grave
- Problemas emocionales y sociales, como la agresión en los niños o la timidez en las niñas
- Problemas con el habla y el lenguaje, especialmente entre los adolescentes masculinos
- El X frágil no tiene cura. Es posible tratar algunos síntomas con terapia educativa, de la conducta o física y medicinas. Obtener tratamiento con anticipación para el X frágil puede ser útil.
SINDROME DE ASPERGER
SINDROME DE ASPERGER
El síndrome de Asperger a menudo se considera una forma de autismo de alto funcionamiento. Esto puede llevar a dificultad para interactuar socialmente, repetir comportamientos y torpeza.
Causas
En 1944, Hans Asperger denominó este trastorno "psicopatía autista". La causa exacta se desconoce, pero es muy probable que una anomalía en el cerebro sea la causa del síndrome de Asperger.
Los factores genéticos pueden jugar un papel, ya que el trastorno tiende a ser hereditario, pero no se ha identificado un gen específico.
El síndrome de Asperger es un trastorno generalizado del desarrollo (TGD) o un trastorno del espectro autísta. La principal diferencia entre el síndrome de Asperger y el trastorno autista es que los niños con el síndrome no tienen retrasos cognitivos o del habla.
La afección parece ser más común en los niños que en las niñas.
Aunque las personas con síndrome de Asperger con frecuencia tienen dificultad a nivel social, muchas tienen una inteligencia por encima del promedio y pueden sobresalir en campos como la programación informática y la ciencia. No presentan retraso en el desarrollo cognitivo, las habilidades para cuidar de sí mismos ni la curiosidad acerca del entorno.
Síntomas
Las personas con síndrome de Asperger se tornan demasiado concentradas u obsesionadas con un solo objeto o tema, ignorando todos los demás. Quieren saber todo sobre este tema y, con frecuencia, hablan poco de otra cosa.
- Los niños con síndrome de Asperger presentan muchos hechos acerca del asunto de su interés, pero parecerá que no hay ningún punto o conclusión.
- Con frecuencia, no reconocen que la otra persona ha perdido interés en el tema.
- Las áreas de interés pueden ser bastante limitadas, como una obsesión con los horarios de los trenes, los directorios telefónicos, una aspiradora o colecciones de objetos.
Las personas con síndrome de Asperger no se aíslan del mundo de la manera en que lo hacen las personas con un trastorno autista. Con frecuencia se acercarán a otras personas. Sin embargo, sus problemas con el habla y el lenguaje en un escenario social a menudo llevan al aislamiento.
- Su lenguaje corporal puede ser inusual.
- Pueden hablar en un tono monótono y pueden no reaccionar a los comentarios o emociones de otras personas.
- Pueden no entender el sarcasmo o el humor, o pueden tomar una metáfora literalmente.
- No reconocen la necesidad de cambiar el volumen de su voz en situaciones diferentes.
- Tienen problemas con el contacto visual, las expresiones faciales, las posturas del cuerpo o los gestos (comunicación no verbal).
- Pueden ser estigmatizados por otros niños como "raros" o "extraños".
Las personas con síndrome de Asperger tienen problemas para formar relaciones con niños de su misma edad u otros adultos, debido a que:
- Son incapaces de responder emocionalmente en interacciones sociales normales.
- No son flexibles respecto a rutinas o rituales.
- Tienen dificultad para mostrar, traer o señalar objetos de interés a otras personas.
- No expresan placer por la felicidad de otras personas.
Los niños con síndrome de Asperger pueden mostrar retrasos en el desarrollo motor y comportamientos físicos inusuales, como:
- Retardo en ser capaces de montar en bicicleta, agarrar una pelota o trepar a un juego
- Torpeza al caminar o realizar otras actividades
- Aleteo de los dedos, contorsiones o movimientos con todo el cuerpo que son repetitivos
Muchos niños con síndrome de Asperger son muy activos y también se les puede diagnosticar trastorno de hiperactividad y déficit de atención (THDA). Se puede desarrollar ansiedad o depresión durante la adolescencia y comienzo de la adultez. Igualmente, se pueden observar síntomas del trastorno obsesivo-compulsivo y un trastorno de tic como el síndrome de Tourette.
SINDROME DE PRADER-WILLI
SINDROME DE PRADER-WILLI
Descripción
El síndrome de Prader-Willi (PWS) es una enfermedad genética caracterizada por obesidad con hipotonía e hipogenitalismo, acromicria y retraso mental.
La hipotonía es severa en la época neonatal, conlleva infecciones respiratorias y problemas de alimentación. La obesidad se inicia entre los 6 meses y los 6 años.
Existe un fenotipo conductual muy característico; en los primeros años de vida son niños alegres y bonachones. En la segunda infancia empiezan los problemas de comportamiento, se vuelven obstinados, tienen tópicos de lenguaje que repiten muy a menudo y son frecuentes los accesos de cólera.
Puede aparecer diabetes no cetógena. El coeficiente intelectual es variable, sin que se pueda descartar la existencia de retraso mental que puede ser variable.
El síndrome de Prader-Willi (PWS) es una enfermedad genética caracterizada por obesidad con hipotonía e hipogenitalismo, acromicria y retraso mental.
La hipotonía es severa en la época neonatal, conlleva infecciones respiratorias y problemas de alimentación. La obesidad se inicia entre los 6 meses y los 6 años.
Existe un fenotipo conductual muy característico; en los primeros años de vida son niños alegres y bonachones. En la segunda infancia empiezan los problemas de comportamiento, se vuelven obstinados, tienen tópicos de lenguaje que repiten muy a menudo y son frecuentes los accesos de cólera.
Puede aparecer diabetes no cetógena. El coeficiente intelectual es variable, sin que se pueda descartar la existencia de retraso mental que puede ser variable.
Consideraciones importantes
Estos pacientes necesitan una atención constante multidisciplinar para intentar paliar las distintas complicaciones que pueden ir apareciendo a lo largo de su vida. Necesitan también apoyo psiquiátrico y/o psicológico, ya que pueden llegar a padecer problemas de autoestima y trastornos de conducta.
La expectativa de vida puede estar reducida debido a las complicaciones de la obesidad, aproximadamente sobre los 35 años. Si se consigue controlar la obesidad y sus complicaciones, esta expectativa de vida mejora considerablemente. Para ello se necesita una dieta estricta, con una disminución de la ingesta de calorías que llega a ser dramática, y un programa de ejercicio muy controlado. Pueden ser niños difíciles y para conseguir un cierto éxito es imprescindible una buena colaboración entre la familia y los facultativos que atienden al paciente.
No debemos olvidar que presentan una disminución de la sensibilidad frente al dolor, lo que dificultará el diagnóstico de algunos problemas.
La expectativa de vida puede estar reducida debido a las complicaciones de la obesidad, aproximadamente sobre los 35 años. Si se consigue controlar la obesidad y sus complicaciones, esta expectativa de vida mejora considerablemente. Para ello se necesita una dieta estricta, con una disminución de la ingesta de calorías que llega a ser dramática, y un programa de ejercicio muy controlado. Pueden ser niños difíciles y para conseguir un cierto éxito es imprescindible una buena colaboración entre la familia y los facultativos que atienden al paciente.
No debemos olvidar que presentan una disminución de la sensibilidad frente al dolor, lo que dificultará el diagnóstico de algunos problemas.
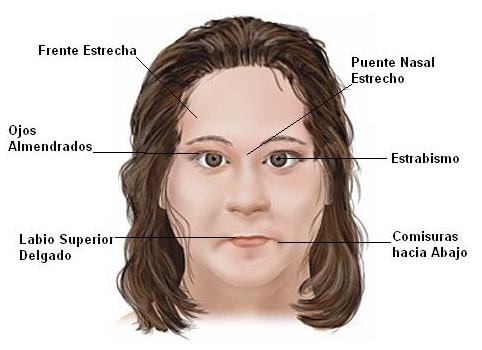
SINDROME DE RETT
Es un trastorno del sistema nervioso que lleva a una regresión en el desarrollo, especialmente en las áreas del lenguaje expresivo y el uso de las manos.
Causas
El síndrome de Rett se presenta casi siempre en las niñas y se puede diagnosticar erróneamente como autismo o parálisis cerebral.
Los estudios han asociado muchos casos de síndrome de Rett con un defecto en un gen de la proteína 2 de unión a metil-CpG (MeCP2). El gen se encuentra en el cromosoma X. Las mujeres tienen dos cromosomas X. Aun cuando un cromosoma presente este defecto, el otro cromosoma es lo suficientemente normal para que la niña sobreviva.
Los varones nacidos con este gen defectuoso no tienen un segundo cromosoma X para compensar el problema, por lo tanto, el defecto generalmente ocasiona aborto espontáneo, mortinato o muerte muy prematura.
La enfermedad afecta a aproximadamente 1 de cada 10,000 niños. Los grupos de la enfermedad han aparecido dentro de familias y ciertas áreas del mundo, como Noruega, Suecia y el norte de Italia.
Síntomas
Un bebé con el síndrome de Rett generalmente tiene un desarrollo normal durante los primeros 6 a 18 meses de vida. Los síntomas varían de leves a graves y pueden abarcar:
- Problemas respiratorios que tienden a empeorar con el estrés; la respiración generalmente es normal durante el sueño y anormal al estar despierto
- Cambio en el desarrollo
- Babeo y salivación excesiva
- Brazos y piernas flácidos (frecuentemente es el primer signo)
- Discapacidades intelectuales y dificultades de aprendizaje (sin embargo, evaluar las destrezas cognitivas en aquellas personas con síndrome de Rett es difícil debido a las anomalías en el lenguaje y el movimiento de la mano)
- Escoliosis
- Marcha temblorosa, inestable o rígida; o caminar sobre los dedos de los pies
- Convulsiones
- El crecimiento de la cabeza se hace más lento comenzando aproximadamente entre los 5 y 6 meses de edad
- Pérdida de los patrones normales de sueño
- Pérdida de los movimientos con propósito de la mano; por ejemplo, el agarre utilizado para recoger objetos es reemplazado por movimientos repetitivos de la mano como torsión de la mano o colocación constante de la mano en la boca
- Pérdida del compromiso social
- Estreñimiento y reflujo gastroesofágico (GERD) continuos y graves
- Circulación deficiente que puede llevar a piernas y brazos fríos y de color azuloso
- Problemas graves en el desarrollo del lenguaje
Suscribirse a:
Entradas (Atom)